
Reproducing proteomics results
at least a part of it and learn SILAC data-analysis on the way...

In the spirit of being FAIR, Findable, Accessible, Interoperable, and Reusable https://www.nature.com/articles/s41597-025-04451-9 i will try to reproduce a finding i was part of i.e. UNG2 depletion https://translational-medicine.biomedcentral.com/articles/10.1186/s12967-020-02318-8 Histone deacetylase inhibitors (HDACi) like “valproate has been used for decades in epilepsy treatment”, so let’s get going 🤞
Data is deposited at publicly accessible proteomics database PRIDE under Histone deacetylase inhibitors mediate selective degradation of nuclear uracil-DNA glycosylase 2 and increased genomic uracil (ebi.ac.uk) so very much Findable 😊
We will use 1st technical replicate for each biological replicate of data Accessible at
http://ftp.ebi.ac.uk/pride-archive/2020/04/PXD008293/151124_JURKAT_SAHA_SILAC_Biol2_tech1.raw
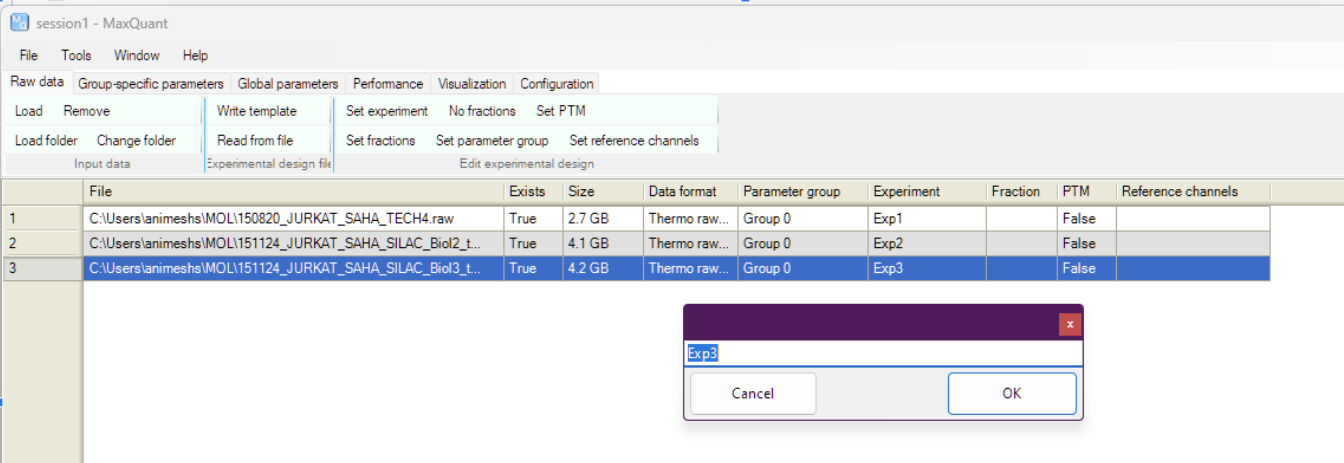
Exp3
http://ftp.ebi.ac.uk/pride-archive/2020/04/PXD008293/151124_JURKAT_SAHA_SILAC_Biol3_tech1.raw
The software used MaxQuant (download https://www.maxquant.org/download_asset/maxquant/latest) is in version 2.6.7.0,
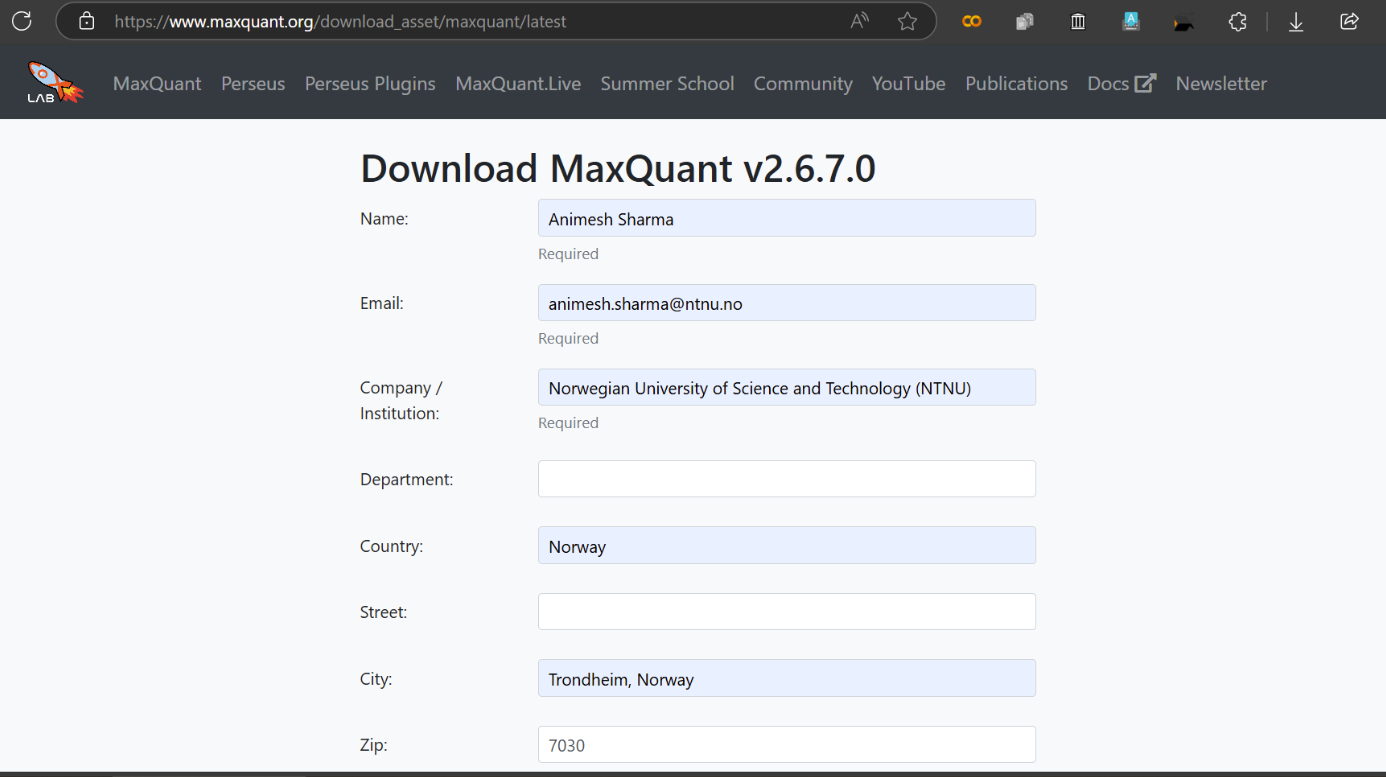
much higher than 1 used in original report but hopefully Interoperable although the parameters need to be recreated so unfortunately not Reusable 🤪 No sweat though, as the defaults are pretty much the same 👍
We will also need the proteome before we can set it up.
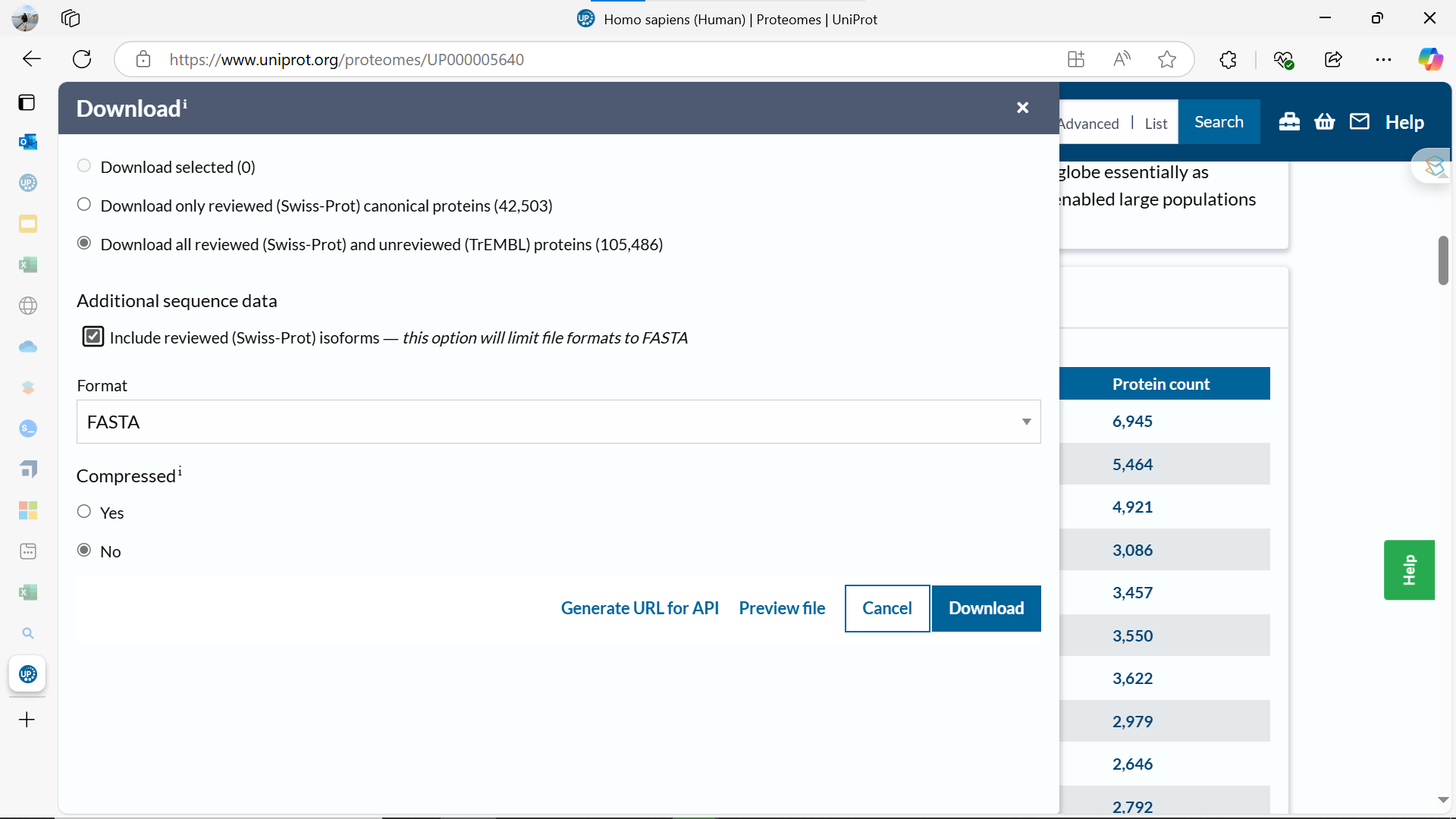
OR just
Now we can setup the search by unzipping the MaxQuant bundle and clicking on “StartMaxQuant.bat”, might get the following warning
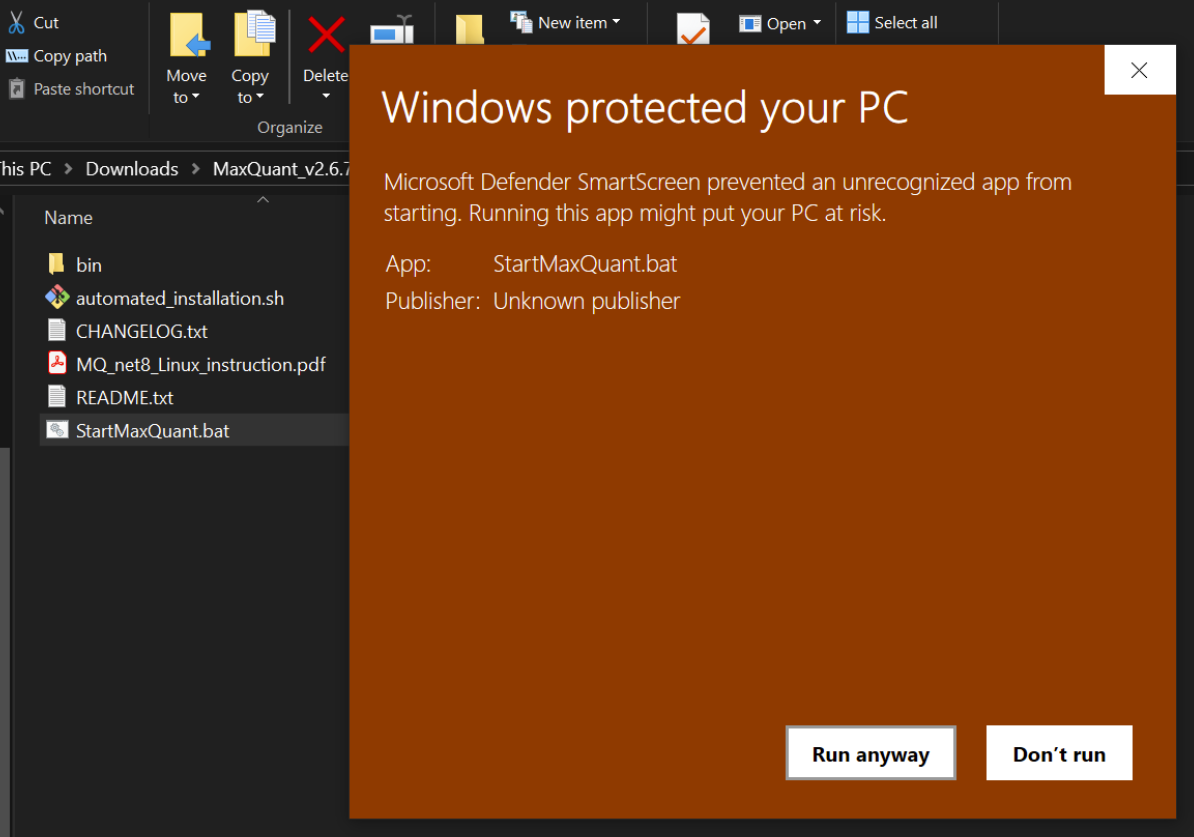
which i just ignore and “Run anyway” 😜
It might ask for dotnet though
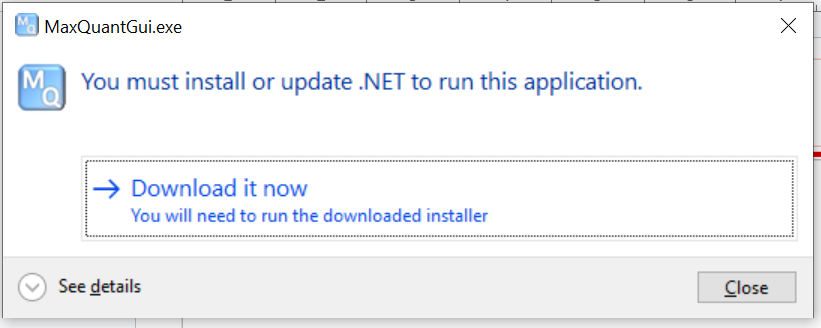
which should download latest version, for me it was https://dotnet.microsoft.com/en-us/download/dotnet/thank-you/runtime-desktop-8.0.13-windows-x64-installer?cid=getdotnetcore
and then you should see the GUI where we need to drag-and-drop the downloaded raw files
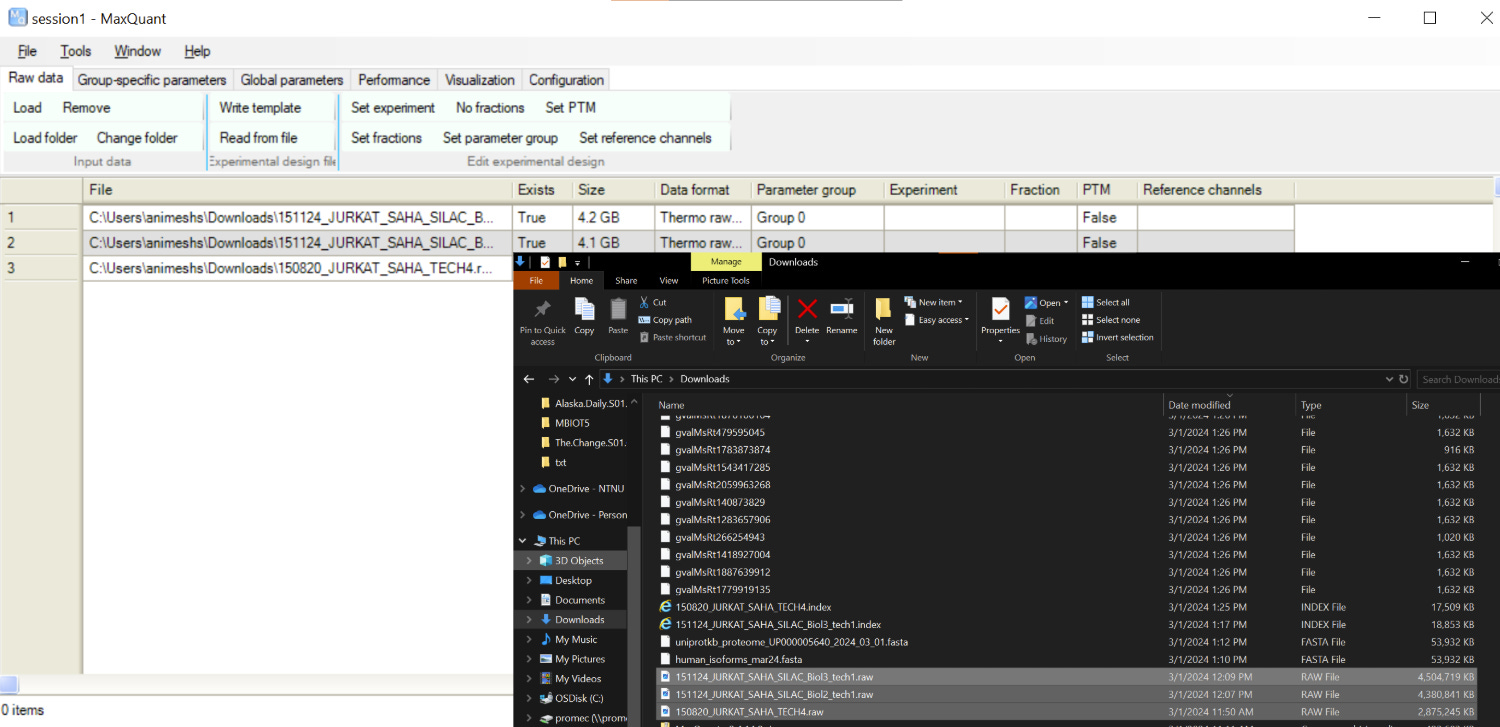
Better to give some meaningful names to the file by selecting the file and clicking on “Set experiment”. This is what is going to be reported in the results, being creative, i will just call them Exp1,2,3 🙃
Now the coolest part is that this is a data coming from an experiment where controls are grown in a medium where Lysine and Arginine are isotopically labelled thus shifting their mass by 8 and 10 Daltons respectively. The cells treated with the HDACi are just as is
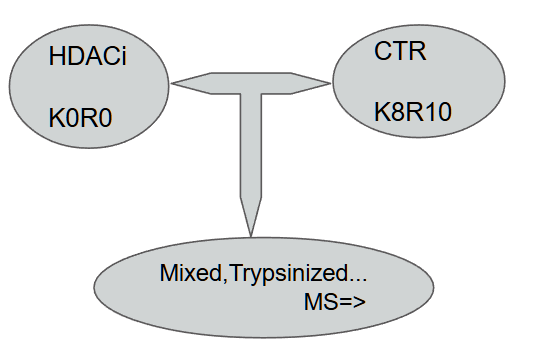
Since they are mixed together 1:1 and everything downstream is same, we consider it as “unbiased comparative study” leading to an awesome batch-effect control! The way we tell this to MaxQuant again is by clicking around in the appropriate boxes
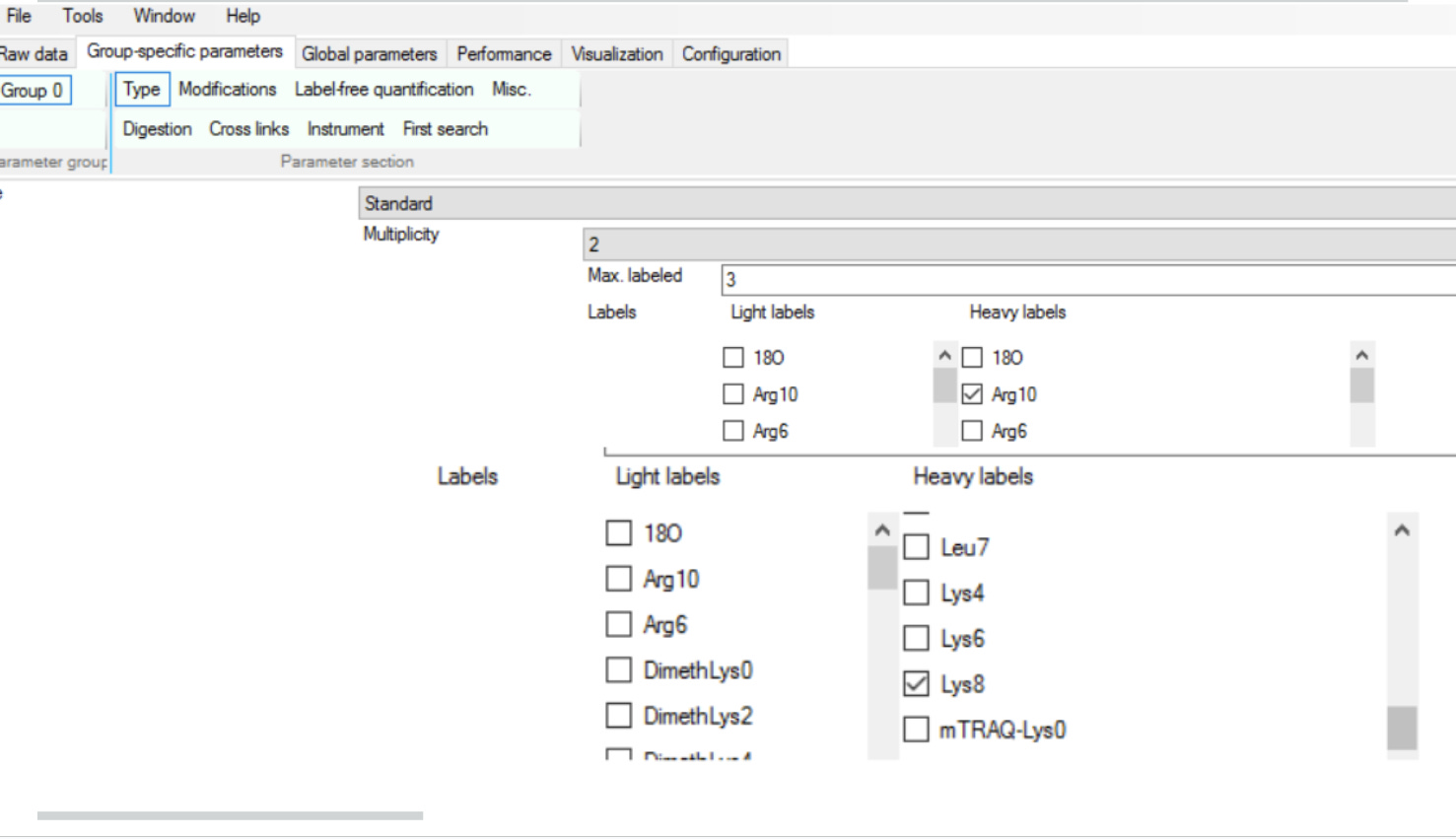
and in case the one of the versions of is not found, MaxQuant will try its best to impute the values, but we have to tell it to do so by clicking “Re-quantify”
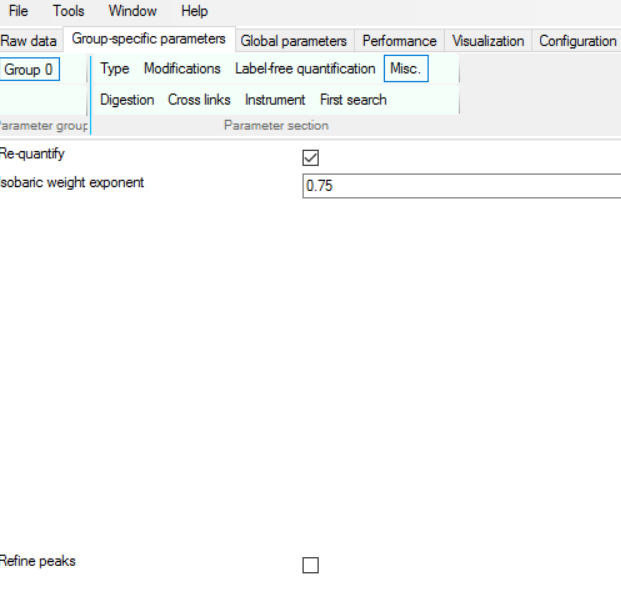
I usually try to maximize the information extraction by reducing the number of peptides to 1 from 2 but make sure it is unique by changing following settings
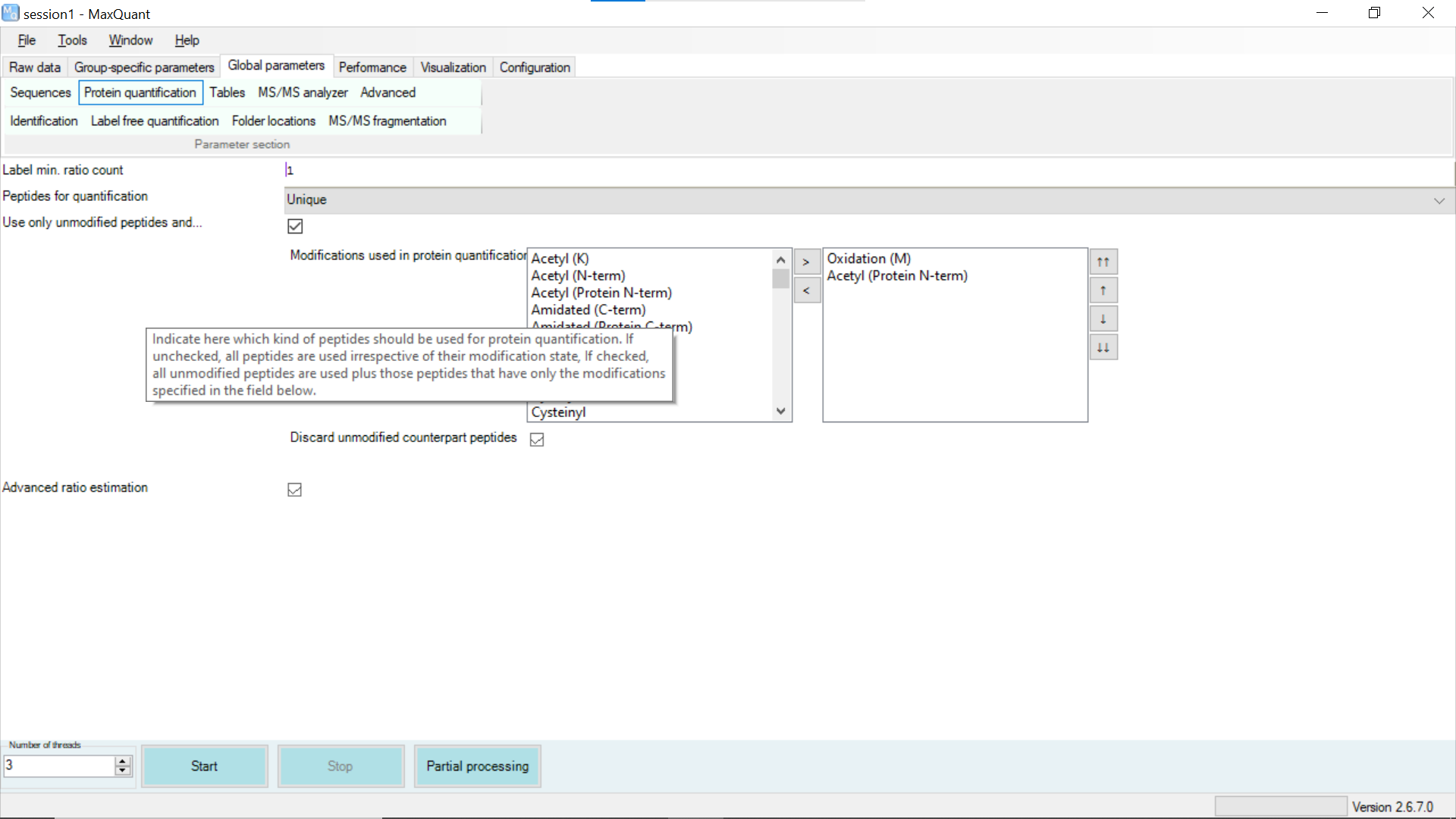
AND
Finally, need to tell MaxQuant about the proteome by dragging the downloaded fasta file into following window
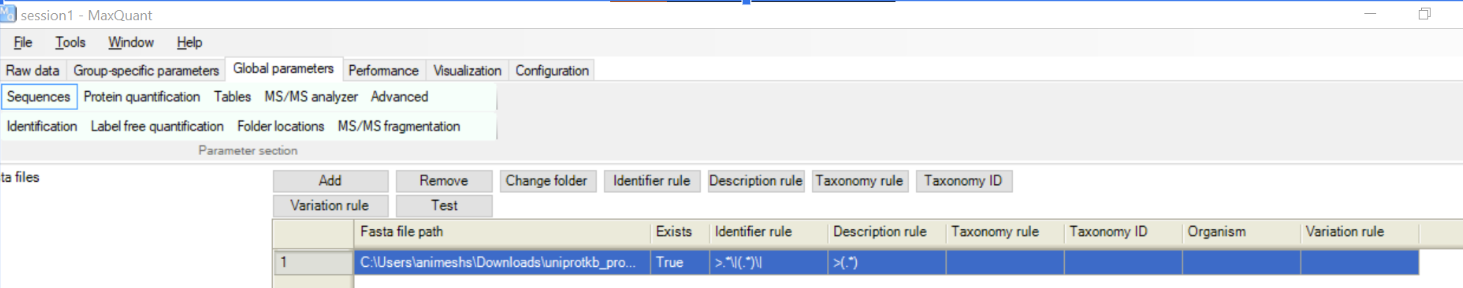
Now just set the number of threads and press “Start”

If all goes well, “Done” popup should appear 🤞
Took about 6 hours with 3 threads on Dell ultrabook!
Will follow up with a post on how to process the results 👍