
RNA-seq analysis with nextflow
bulk transcriptomics as simple as it can get without compromising quality

Details of instructions are available at https://animesh.github.io/rnaseq_nextflow/ which essentially consists of creating a directory for the analysis or simply clone this repo, download raw data create sample-sheet from the downloaded data, download Reference Genome and the corresponding annotation for the Reference Genome, install nextflow (needs curl, Java and Docker or Singularity on Linux beforehand (Windows users can run via WSL) and finally run it over the downloaded data above using the created sample-sheet, reference genome and the corresponding annotation
curl -s https://get.nextflow.io | bash
./nextflow run nf-core/rnaseq --max_memory '16.GB' --max_cpus 6 --input samples.csv --outdir results --gtf chr22_with_ERCC92.biotype.gtf --fasta chr22_with_ERCC92.fa -profile docker This should create results folder containing nextflow multiqc_report
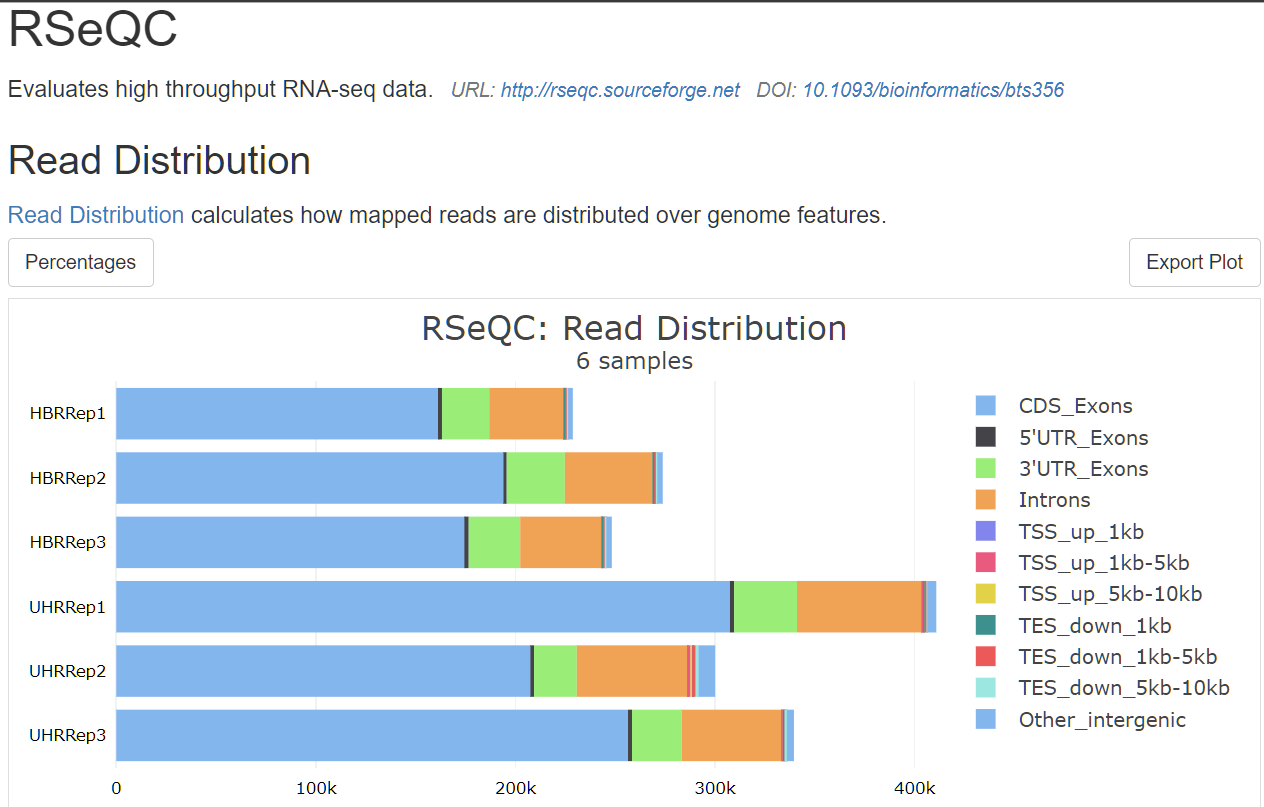
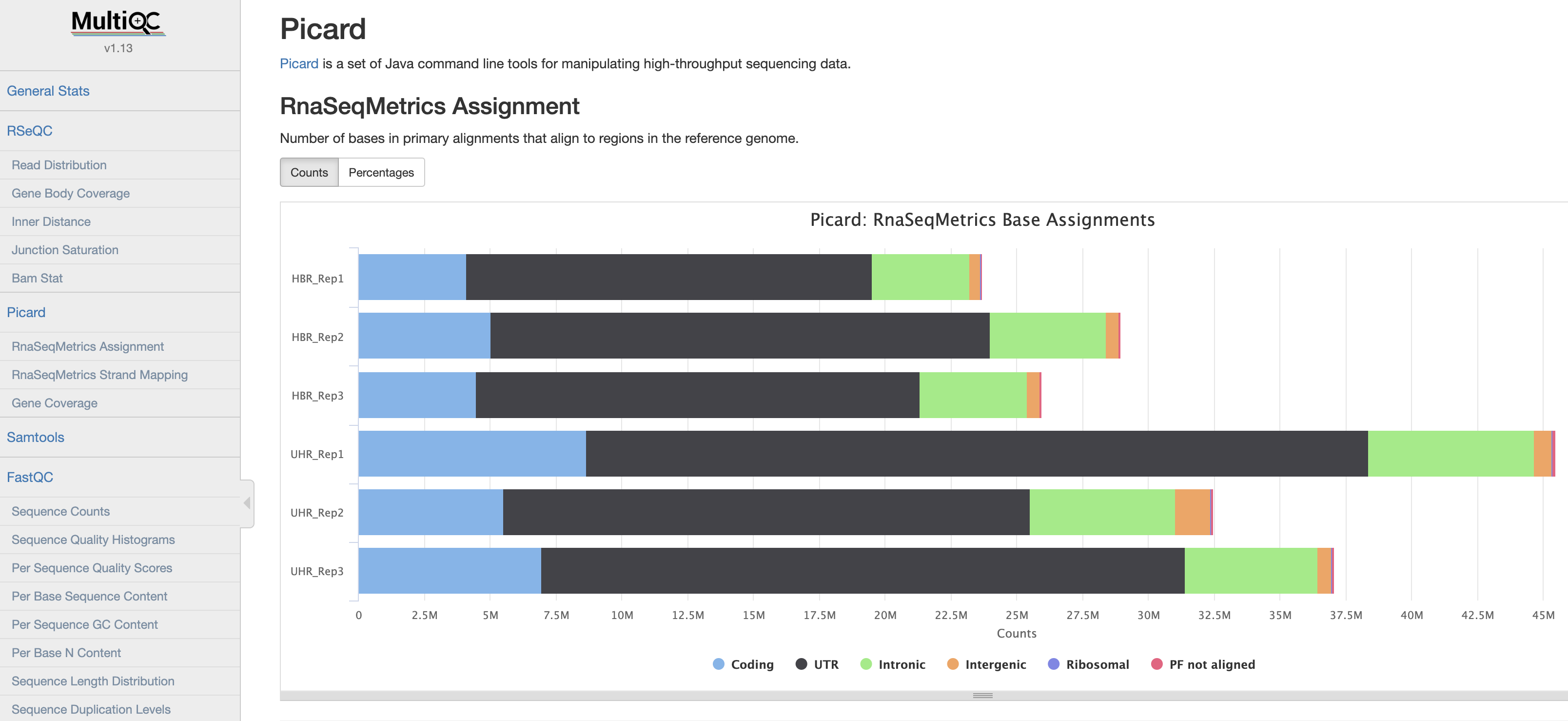
which does looks similar to original multi-QC-report
{kind=link}
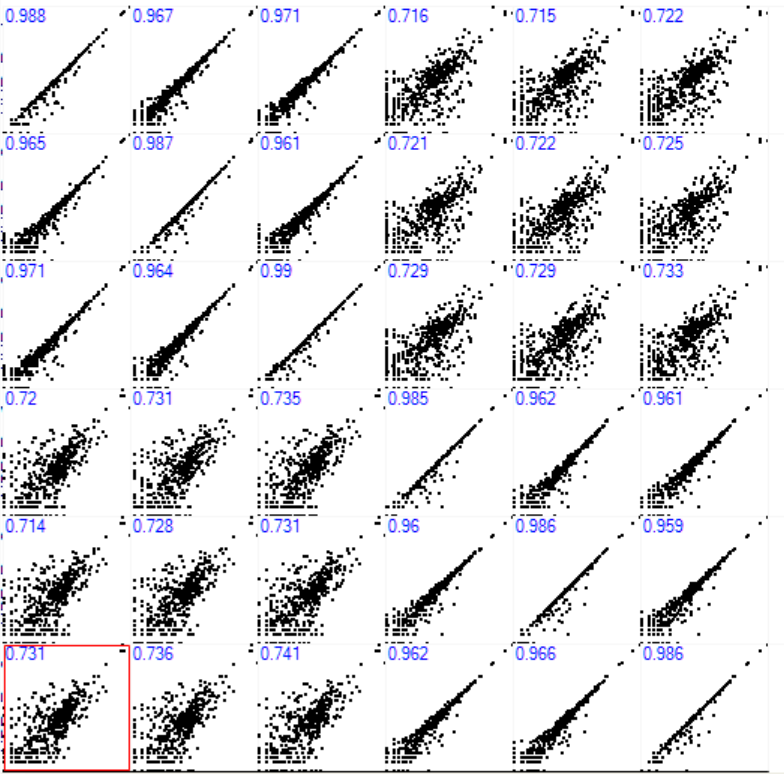
Coming to the actual Result , specifically the gene-counts from original analysis and comparing with resulting nextflow-gene-counts, spearman rank-correlation seems to be about ~ 0.99 calculated using Perseus shown in blue in scatter-plot
{kind=link}

where the x-axis represents the nextflow and it looks like it usually has higher or similar count as the original result, looking into the euclidean-distance clusters
{kind=link}
")
between the samples counts (scale in 10E4) so it seems like nextflow pipeline has performed quite well as original if not better, all in a command-line!
Will look deeper into the results and probably run the differential-expression analysis pipeline, do keep an eye 🤞